Qu'est-ce que le facteur von Willebrand ?
Le facteur von Willebrand est une protéine présente dans la circulation sanguine. Elle y est libérée par les plaquettes sanguines d'une part et les cellules qui revêtent les vaisseaux sanguins d'autre part. Le facteur von Willebrand possède de multiples fonctions et présente une structure très complexe.
Chaque facteur von Willebrand est en effet constituée de 20 à 50 sous-unités identiques liées les unes aux autres comme les maillons d'une chaîne, ce qui en fait la plus grande protéine présente dans notre sang.
Chaque maillon de la chaîne possède des points de contacts ou récepteurs permettant au facteur von Willebrand d'interagir avec les plaquettes d'une part et la paroi lésée des vaisseaux sanguins d'autre part. Un autre récepteur permet au facteur von Willebrand de lier et de protéger un autre facteur important de la coagulation sanguine, le facteur VIII. C'est ce facteur qui fait défaut dans une forme d'hémophilie appelée hémophilie A.
Pour assurer ses fonctions, le facteur von Willebrand doit être présent en quantités suffisantes dans le sang et posséder une structure normale lui permettant de fixer les plaquettes, les parois de vaisseaux et le facteur VIII de la coagulation sanguine.
Pourquoi la maladie de von Willebrand s'accompagne-t-elle de saignements ?
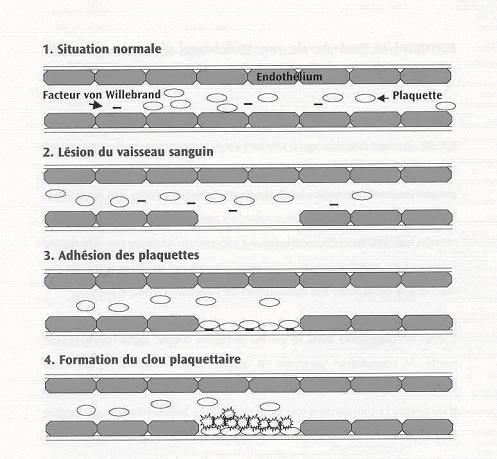
Notre sang circule dans un circuit fermé de vaisseaux sanguins. En cas de blessure, une brèche est créée dans le vaisseau sanguin, par laquelle le sang peut s’échapper. Pour empêcher cela, la première mesure sera de colmater cette brèche en attirant les plaquettes sanguines, entraînant ainsi la formation d’un « clou plaquettaire » (amas formé de plaquettes sanguines). Le saignement est alors arrêté (tout au moins provisoirement). Ce processus, appelé « hémostase primaire », correspond à la première phase de la coagulation. Ce clou plaquettaire ne peut pas résister longtemps à la pression sanguine ; il doit donc être renforcé par la formation d’un réseau de fibrine (protéine de coagulation). Pour ce faire, les protéines de coagulation (facteurs) sont activées une à une, en cascade, pour aboutir finalement à la formation de fibrine. La plupart de ces protéines de coagulation sont désignées par un numéro, et certaines portent également un nom particulier. Il s’agit de la seconde phase de coagulation, appelée « hémostase secondaire » ou « coagulation plasmatique ». Le réseau de fibrine transforme le clou plaquettaire en caillot sanguin à part entière, lequel peut rester longtemps en place, jusqu’à réparation complète du vaisseau sanguin.
Le facteur von Willebrand (FvW) incite les plaquettes sanguines à s’agglutiner dans la brèche du vaisseau sanguin, tant sur la paroi du vaisseau qu’entre elles. En l’absence du facteur von Willebrand (FvW), il est donc impossible d’obtenir un bon clou plaquettaire. Si le FvW est présent en moindre quantité ou s’il fonctionne moins bien, les saignements dureront plus longtemps et les pertes de sang seront plus importantes.
Le facteur de coagulation VIII est un facteur « faible », car il ne peut pas survivre longtemps dans la circulation sanguine. Le FvW est une grosse protéine qui se lie à ce facteur VIII ; le facteur VIII est ainsi protégé et peut donc rester plus longtemps présent dans la circulation. S’il y a moins de FvW, il y a donc aussi moins de facteur VIII, et donc moins de formation de fibrine. Lorsque la fibrine se forme plus lentement ou en moindre quantité, le clou plaquettaire est moins solide. Le saignement durera plus longtemps ou le patient recommencera à saigner.
La maladie de von Willebrand se caractérise donc par un dysfonctionnement dans les deux phases de la coagulation :
• la phase liée à la formation du clou plaquettaire
• la phase liée à la formation du réseau de fibrine.